Proteogenomic Proof: Using Mass Spectrometry to Confirm and Correct Gene Models with ProteoAnnotator
Proteogenomics uses mass-spectrometry (MS) peptide evidence to show which predicted gene models are actually translated, and tools such as ProteoAnnotator provide a reproducible pipeline to map peptide identifications back to the genome and score proposed annotation changes.
Why proteogenomics matters (short primer)

Genome annotation pipelines predict genes from sequence and transcript evidence, but predictions can be wrong at exon boundaries, start sites, splice isoforms, or entire coding regions. MS-based proteomics gives direct evidence of translation: if a peptide uniquely maps to a predicted coding sequence, that model is supported; if peptides map to regions outside annotations, they flag missing or mis-annotated genes. Integrating proteomic evidence raises annotation confidence and helps convert predicted open reading frames (ORFs) into validated protein products.

ProteoAnnotator
what it is and what it does

ProteoAnnotator is an open-source proteogenomics annotation framework designed to:
Accept peptide identifications (e.g., mzIdentML) from common search engines.
Combine multiple search results and custom FASTA databases.

Compute peptide- and protein-level statistics.
Map peptides back to genomic coordinates
Provide locus-level scoring that supports recommended edits to gene models
Design the proteogenomic search database
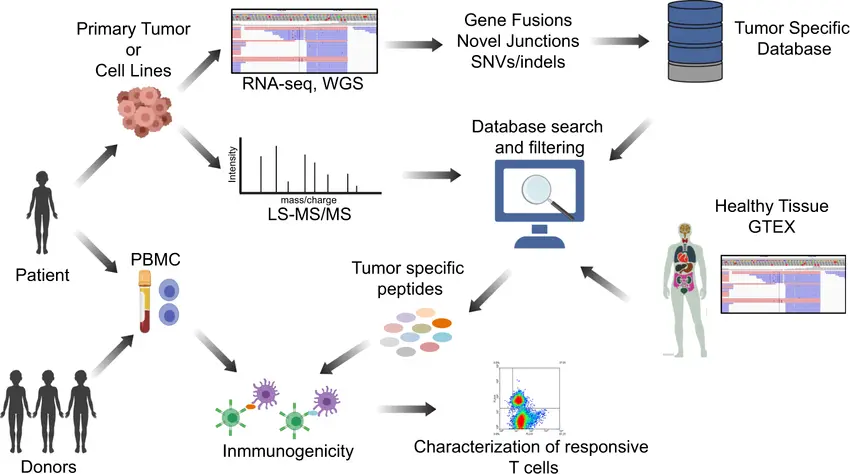
Build a FASTA that contains: the canonical proteome (reference proteins), translated products from predicted gene models (including alternative splices and 6-frame translations where appropriate), and variant/novel ORFs derived from RNA-seq. Use well documented mapping files so peptide→genome relationships are traceable.
Sample preparation & MS acquisition
Use deep coverage bottom-up proteomics (fractionation, long gradients) to maximize unique peptide detection. If isoform resolution is critical, consider targeted workflows or top-down strategies for intact proteoform evidence.
Database search & FDR control
Search raw data against the augmented FASTA with one or more search engines. Carefully control false discovery rate (FDR) at peptide and protein levels proteogenomics often uses more stringent thresholds because the search space is expanded. Modular pipelines (combining engines and rescoring) improve sensitivity while controlling FDR.
Peptide→genome mapping
Translate peptide coordinates into genomic positions (accounting for frame and splice junctions). ProteoAnnotator and similar tools provide mapping modules that attach peptide evidence to loci.
Locus scoring and annotation decision
Instead of treating every peptide equally, compute locus-level scores that consider: number of unique peptides, peptide uniqueness across isoforms, spectrum quality, and consistency with transcript evidence. ProteoAnnotator implements grouped scoring and statistical tests to decide whether a gene model needs correction.
Manual curation & integration
High-confidence loci are then inspected by curators with genome browsers (showing peptides, transcripts, and gene models). Proposed changes are added to annotation databases with provenance (which peptide, which spectrum, FDR, etc.).

What MS evidence can (and cannot) prove
Can prove (robust):
- A predicted exon is translated when one or more uniquely mapping peptides cover it.
- Splice junctions when peptides span exon–exon junctions (requires appropriately designed search databases).
- Protein expression in a tissue/sample and presence of specific isoforms if unique peptides are observed.
Limited or ambiguous:
- Small proteins/short ORFs (<7–8 aa peptide coverage) are hard to detect reliably with standard bottom-up proteomics.
- Absence of peptide evidence is not proof of absence low expression, proteolytic properties, or MS sampling depth can cause false negatives.