Start writing here...
in News

Why Validate Gene Models?
1
Ensure coding sequence integrity
Predicted models may contain frame shifts or miss start/stop codons.
2
Confirm splicing events
Alternative exons and junctions require experimental validation.
3
Avoid false positives
Some computational predictions annotate pseudogenes as coding.
4
Support functional annotation
Validated proteins provide higher confidence for downstream biological interpretation.
The Role of ProteoAnnotator
ProteoAnnotator was developed by the HUPO Proteomics Standards Initiative (PSI) to unify proteogenomic analyses into a reproducible and standards-driven framework. Its strengths include:
Supports mzIdentML inputs from search engines such as MS-GF+, X!Tandem, and Mascot.
Integration of multiple search results
Supports mzIdentML inputs from search engines such as MS-GF+, X!Tandem, and Mascot.
Mapping of peptides to genome coordinates
Produces proBed and proBAM files for visualization in IGV or JBrowse.
Standardized outputs
mzIdentML ensures interoperability across pipelines, while proBed/proBAM enable community data sharing.
Detection of annotation discrepancies
Identifies peptides mapping outside known coding regions, suggesting novel exons or isoforms.

Input Preparation
- Genome annotation: GFF/GTF of existing gene models.
- Protein FASTA: Translations of annotated CDS regions.
- MS data: Raw files converted to mzML, followed by peptide identifications (mzIdentML).
- Optional transcriptome: RNA-seq BAM or GTF files for cross-validation.

Database Searching
MS/MS spectra are searched against the protein FASTA, with decoys included for FDR estimation. The resulting mzIdentML captures peptide-spectrum matches (PSMs).
Peptide Mapping with ProteoAnnotator
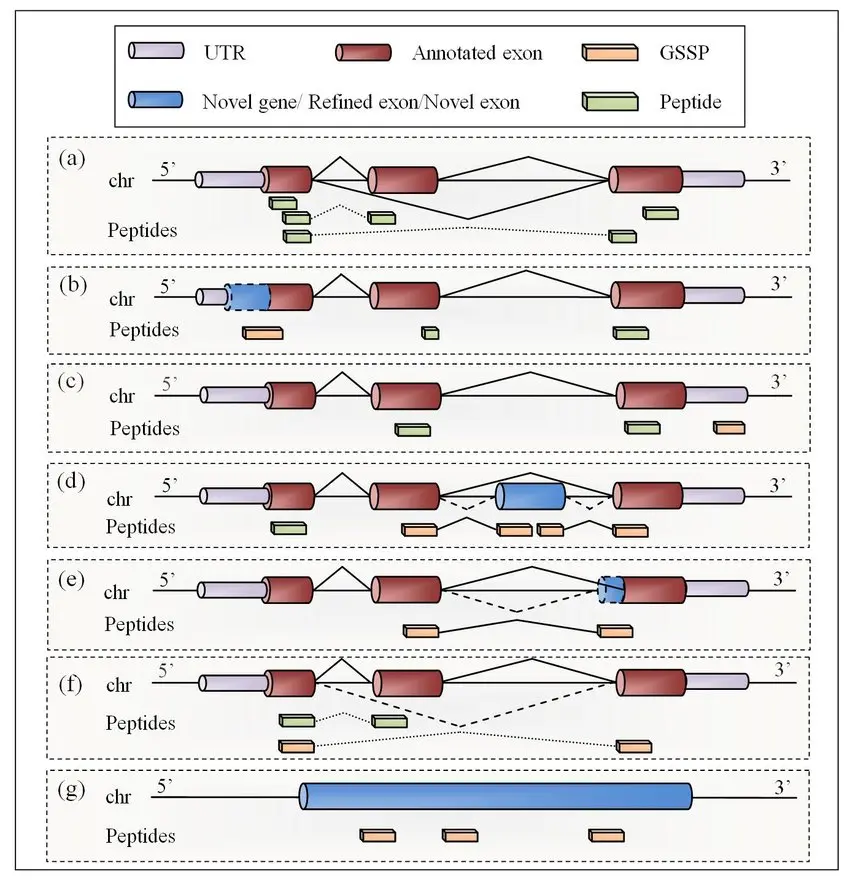
ProteoAnnotator aligns validated peptides to annotated proteins and their genomic coordinates. This step highlights:
- Supported exons: Peptides that confirm annotated regions.
- Splice junction peptides: Direct evidence of exon-exon boundaries.
- Novel peptides: Evidence of unannotated exons, frameshifts, or alternative isoforms.
Quality Control
- PSM FDR ≤ 1%.
- Minimum of 2 unique peptides per protein for strong validation.
- Manual review of single-peptide identifications, ideally supplemented by RNA-seq support.
Visualization and Interpretation
The proBed/proBAM outputs are loaded into genome browsers. Researchers can visually inspect how peptide evidence aligns with annotated CDS, splice junctions, or unannotated regions.