1.ProteoAnnotator
ProteoAnnotator is a completely automated software pipeline for integration of mass spectrometry (MS) based proteomic evidence into genome databases. The goal is to use MS data for confirming that predicted gene models are translated into protein, and to verify the correctness of the existing gene models. It is also used to examine whether there is supporting evidence for alternative gene predictions at particular loci.
Its primary function is to provide experimental evidence for the validation and refinement of predicted gene models. Below is a technical breakdown of its features and relevance to biotechnology:
1
Gene Model Validation
ProteoAnnotator verifies if predicted genes in a genome are expressed as proteins. This is done by matching peptides identified through MS experiments to the corresponding genomic regions.
- Purpose: Ensures the accuracy of gene predictions.
- Output: Confirms expressed proteins or flags incorrect models.
2
Alternative Gene Predictions
The tool identifies proteomic evidence supporting variations in gene models, such as alternative splicing or additional exons.
- Functionality: Detects peptides that align with alternative gene isoforms.
- Use Case: Improves understanding of gene structures and their functional diversity.
3
Novel Protein Identification
ProteoAnnotator can detect new proteins that were not included in initial genome annotations.
- Application: Helps discover genes or coding regions missed during computational predictions.
- Outcome: Expands genome annotations with experimentally verified information.
4
Refining Genome Annotations
ProteoAnnotator aids in updating genome databases by:
- Adjusting gene boundaries based on proteomic data.
- Providing evidence for untranslated regions (UTRs).
- Identifying errors in exon-intron assignments.
5
Post-Translational Modifications
The software detects modifications like phosphorylation and glycosylation in proteins.
- Purpose: Adds functional context to proteins by highlighting biochemical changes.
- Significance: Helps annotate proteins with roles in metabolic pathways or regulatory networks.
6
Automated Data Integration
ProteoAnnotator automates the process of comparing MS data with genomic sequences.
- Efficiency: Reduces manual errors and speeds up analysis.
- Scalability: Suitable for high-throughput research projects.
7
Cross-Species Applications
The tool allows comparison of proteomic data across different organisms.
- Application: Identifies conserved proteins and genes.
- Benefit: Enhances the annotation of less-characterized genomes using data from model organisms.
8
Quality Control
ProteoAnnotator includes checks to ensure accurate mapping of peptides to genes.
- Error Reduction: Flags mismatches and ensures consistent data alignment.
- Confidence Scoring: Assigns reliability scores to protein identifications.
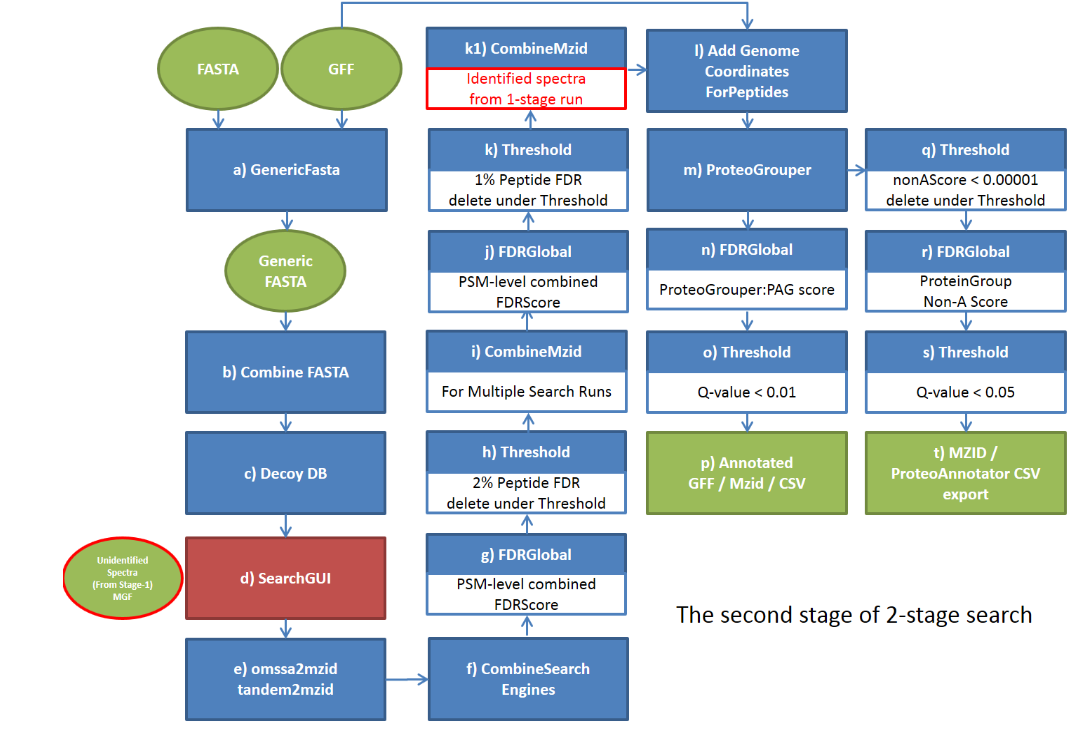
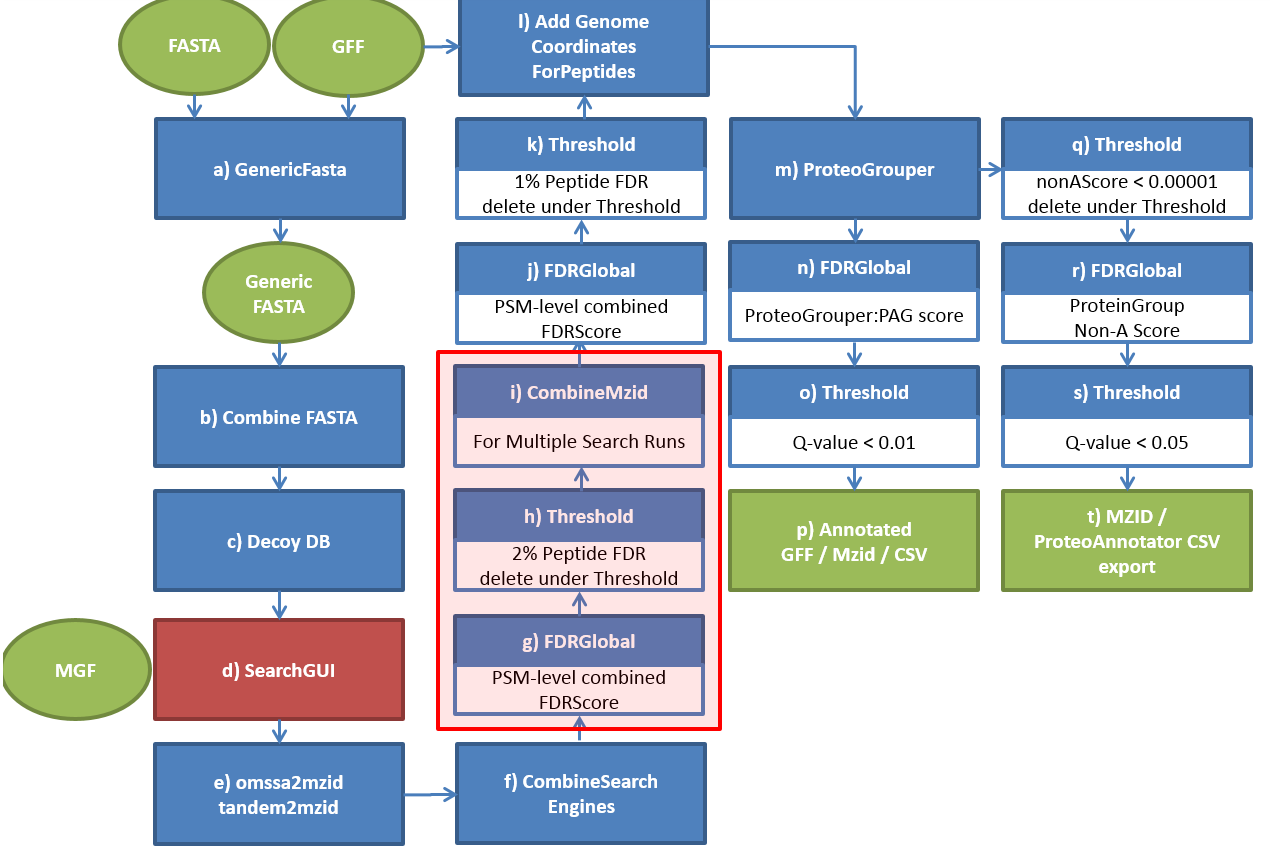
2.Two-Stage Search in ProteoAnnotator
ProteoAnnotator uses a two-stage search method to integrate mass spectrometry (MS) data with genome annotations. This method improves efficiency and accuracy when identifying peptides and validating gene models. Below is a technical description of how this process works:
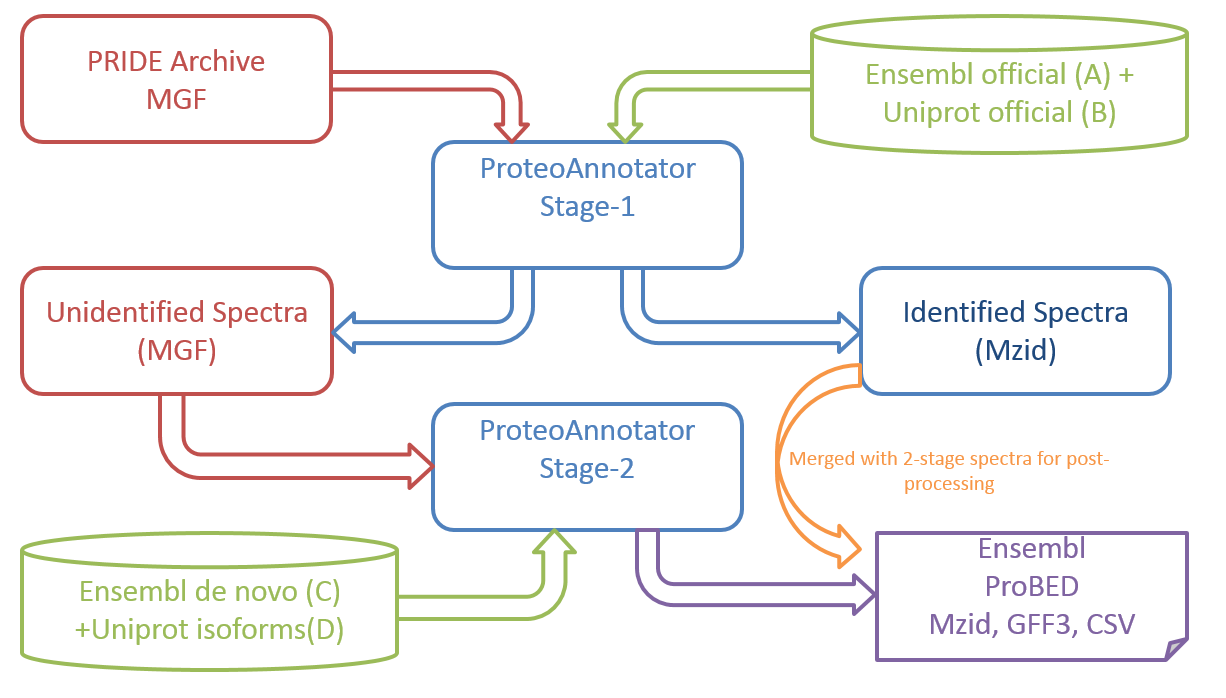
Stage 1: Identified MS Spectra
In this stage, the dataset is rapidly reduced by identifying spectra that could correspond to peptides.
Steps:
- Spectral Matching: MS spectra are compared with theoretical peptide masses derived from predicted gene sequences.
- Tolerances: Broad mass tolerances are applied to ensure that potential matches are not missed.
- Elimination: Spectra that do not match are removed to streamline the dataset.
Output: A condensed list of spectra likely matching peptides.
Efficiency: This stage prioritizes speed, enabling fast processing of large MS datasets.
Stage 2: Validation and Refinement
This stage refines the results from Stage 1 to ensure accurate peptide identification.
Steps:
- Recalibration: Mass measurements are adjusted to enhance precision and reduce errors.
- Detailed Matching: The spectra are re-analyzed using stricter mass tolerances for more accurate peptide alignment.
- Probability Scoring: Algorithms are applied to evaluate the likelihood of a match.
- Post-Translational Modifications (PTMs): Identifies PTMs like phosphorylation or acetylation.
- Cross-Validation: Matches are compared with other experimental or genomic data for verification.
Output: A validated list of peptide matches with accurate annotations.
Accuracy: The focus is on high specificity to minimize false positives.